CRISPR Bacterial Genome Editing Service
CRISPR has rapidly become the genome editing tool of choice in many eukaryote organisms, however its use in bacteria has been limited until recently. Many bacteria do not possess robust DNA repair systems as eukaryotes do, and therefore CRISPR-induced double-stranded DNA breaks are lethal. To improve recombination rates scientists have employed phage-derived (Lambda Red) recombinases to carry out enhanced homologous recombination – referred to as recombineering. When Lambda Red recombination is coupled with CRISPR it provides a powerful tool for selecting against non-edited cells, allowing for highly precise and efficient scarless genome editing. Bacteria possess great potential to produce complex molecules with applications in therapeutics, agriculture, and industry. E. coli has long been a preferred choice for metabolic engineering and the production of such molecules due to its well-characterization and abundance of available genetic tools. Our custom bacterial gene knockout and knock-in services offer highly specific and efficient editing of the E. coli genome. When the locus of your knock-in isn’t a concern, consider our E. coli Safe Harbor Gene Knock-In Service. Our expert scientists have verified this “safe harbor” or “hotspot” locus in the E. coli genome for safe guarded genomic expression. Use this service for reliable expression of your gene of interest. To order bacterial genome editing services, simply send us the information on your desired gene knock-in/knockout and E. coli strain, and we’ll do the rest!
Features:
- CRISPR-assisted gene knockout: Partial or whole gene deletion of your choice.
- CRISPR-assisted gene knock-in: Tag an existing gene or knock-in a gene of interest.
- Tried-and-true genome editing from expert scientists: see our Bacterial CRISPR Case Studies for more information.
- Validated Safe Harbor locus allows for stable, single copy genomic expression of your gene or construct of interest.
Service Details
| E. COLI GENE KNOCKOUT SERVICE | DELIVERABLES | LEAD TIME | CAT. NO. |
|---|---|---|---|
|
Sequence verified engineered strain in glycerol stock (2 different clones) | 8 weeks | C424 |
| E. COLI GENE KNOCKOUT SERVICE | DELIVERABLES | LEAD TIME | CAT. NO. |
|---|---|---|---|
|
Sequence verified engineered strain in glycerol stock (2 different clones) | 9 weeks | C425 |
| E. COLI GENE KNOCKOUT SERVICE | DELIVERABLES | LEAD TIME | CAT. NO. |
|---|---|---|---|
|
Sequence verified engineered strain at the safe harbor locus in a glycerol stock (2 different clones) | 9 weeks | C520 |
Additional Info
Workflow
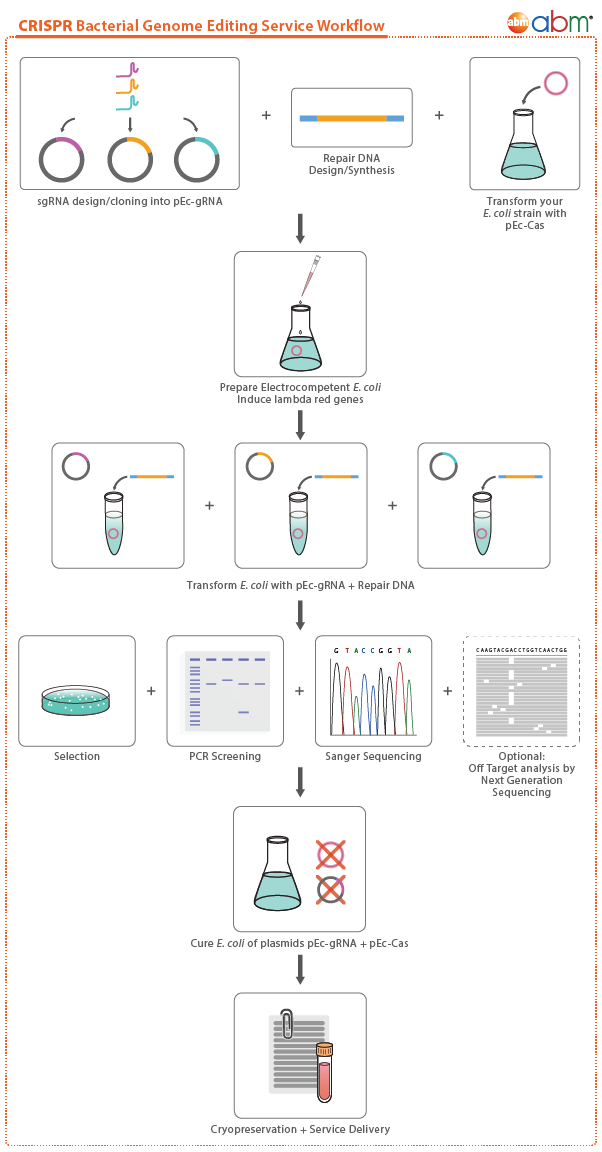
Bacterial Gene Knockout Case Study
CRISPR-assisted knockout of chloramphenicol resistance cassette (CAT) in E. coli.
- A genomically encoded chloramphenicol resistance cassette (referred to as CAT-chloramphenicol acetyl transferase) was knocked out using CRISPR-assisted genome editing.
- E. coli transformants were screened for sensitivity to chloramphenicol and correct chromosomal insertion of repair template.
- CAT knockout was confirmed by sequencing.
Phase #1: Cas9 and sgRNA Design and Cloning
- To improve recombination rates in bacteria, phage-derived (λ red) recombinases were employed alongside Cas9 in pCas to carry out enhanced homologus recombination (Figure 1).
- sgRNAs were designed against the CAT gene which was previously introduced into the E. coli genome at the yeeR locus (accession number: NP_416505). The resulting sgRNAs were then cloned into pTarget (Figure 1).
- Repair templates were designed as single-stranded oligonucleotides containing homology to the CAT gene. The repair template also contains three stop codons for the early termination of CAT and a unique restriction site for screening purposes (Figure 3). Importantly, the repair template eliminates the PAM site, preventing Cas9 re-targeting and cleavage of edited cells.
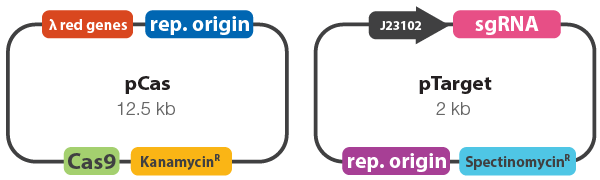
Figure 1. Vector maps of pCas and pTarget. pCas9 constitutively expresses Cas9, whereas the λ red genes are inducible. pTarget constitutively expresses the sgRNA to guide Cas9 to the target locus.
Phase #2: Preparation of λ Red-induced Electrocompetent Cells and Transformation
- pCas, carrying the λ red genes and Cas9, was transformed into E. coli cells. These cells were then made electrocompetent and the λ red genes were induced prior to co-transformation of pTarget and the repair template.
Phase #3: Screening and Sequencing - Knockout of Genomically-encoded Chloramphenicol Resistance Cassette (CAT)
A) Screening for sensitivity to chloramphenicol
- Transformants were replica picked onto kanamycin and chloramphenicol agar plates to assess sensitivity to chloramphenicol.
- Successful knockout and inhibition of the CAT gene is indicated by growth on kanamycin plates, but no growth on chloramphenicol plates (Figure 2).
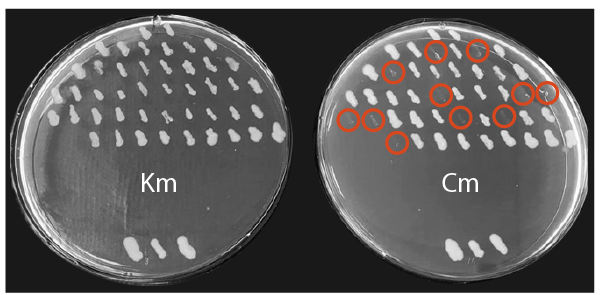
Figure 2. Replica plates of potential CAT knockouts. Replica plates demonstrate 11/45 transformants were successfully edited (circled in red). The kanamycin plate is shown on the left, and the chloramphenicol plate on the right. Wild type controls (carrying the kanamycin plasmid and CAT gene integrated into the chromosome) are shown at the bottom of each plate.
B) Screening for correct chromosomal insertion of repair template by restriction digest
- Successfully edited transformants can be verified by restriction enzyme digest using the unique SpeI site (Figure 3).
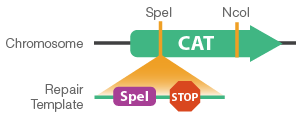
Figure 3. Schematic of CAT gene knock out using a repair template containing stop codons and SpeI site.
- The target locus was PCR amplified from the chloramphenicol-sensitive colonies and then digested using SpeI and NcoI to reveal a unique digest profile (Figure 4).
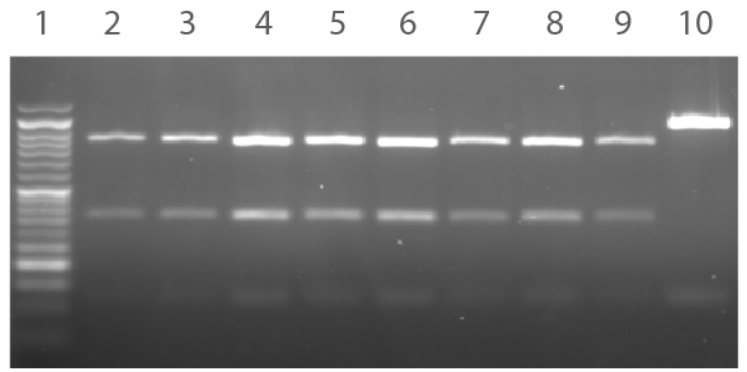
Figure 4. Agarose gel depicting restriction digest profiles of the chloramphenicol- sensitive colonies. PCR products subjected to SpeI/NcoI restriction digest produces three bands for a positive clone and two bands for a negative clone. Lane 1: 100 bp Opti-DNA Marker. Lane 2-9: Colonies #1-8. Lane 10: Negative control.
C) Sequencing of Chloramphenicol-sensitive and Restriction Digest Positive Colonies
- PCR products were subjected to Sanger sequencing to confirm correct insertion and knock out of the CAT gene (Figure 5).
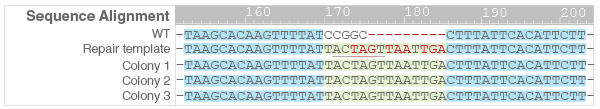
Figure 5. Sequence alignment of CAT gene knockout colonies compared to wild type and repair template. The knockout insertion sequence (green) depicts the three stop codons (red) and the SpeI restriction site (underlined).
Bacterial Gene Knock-In Case Study
CRISPR-assisted knock-in of the mCherry cassette in E. coli.
- The mCherry cassette was knocked into the chromosome using CRISPR-assisted genome editing.
- Transformants were screened using colony PCR.
- mCherry knock-in was confirmed by sequencing.
Phase #1: Cas9 and sgRNA Design and Cloning
- To improve recombination rates in bacteria, phage-derived (λ red) recombinases were employed alongside Cas9 in pCas to carry out enhanced homologus recombination (Figure 1).
- sgRNAs were designed against the yeeR locus (accession number: NP_416505). Each sgRNA was individually cloned into pTarget (Figure 1).
- Repair templates were designed as double-stranded DNA containing the mCherry cassette flanked by homologies to the yeeR locus (Figure 2).
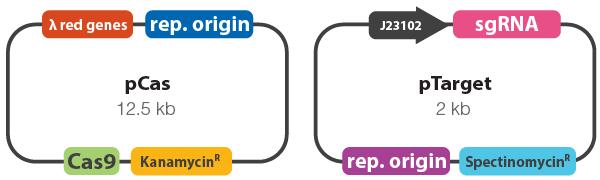
Figure 1. Vector maps of pCas and pTarget. pCas9 constitutively expresses Cas9, whereas the λ red genes are inducible. pTarget constitutively expresses the sgRNA to guide Cas9 to the target locus.
Phase #2: Preparation of λ Red-induced Electrocompetent Cells and Transformation
- pCas, carrying the λ red genes and Cas9, was transformed into E. coli cells. These cells were then made electrocompetent and the λ red genes were induced prior to co-transformation of pTarget and the repair template.
Phase #3: Screening and Sequencing - Knock-in of a Chromosomal mCherry Cassette
A) Colony PCR Screening for Insertion of Chromosomal mCherry Cassette
- Colonies were subjected to PCR using one primer specific to the upstream region of the integration site on the chromosome and one primer specific to the mCherry cassette (Figure 2).
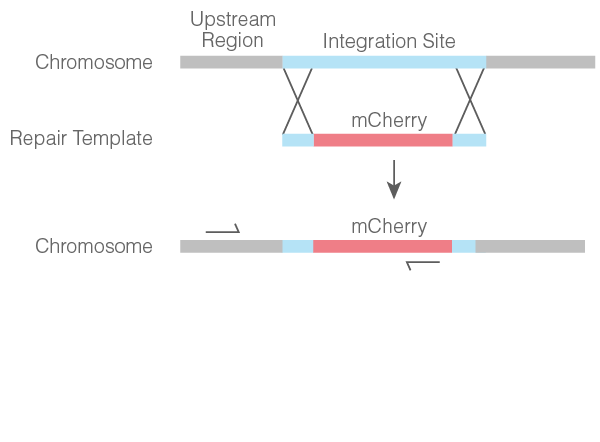
Figure 2. Schematic of mCherry cassette chromosomal knock-in and location of specific primers for colony PCR screening.
- The resulting PCR products were run on an agarose gel to confirm correct chromosomal insertion (Figure 3).
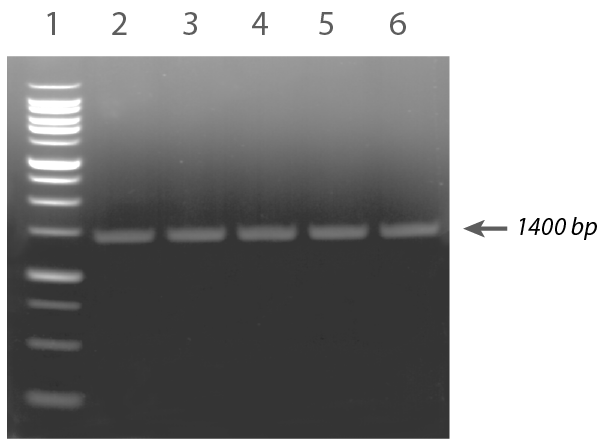
Figure 3. Agarose gel depicting colony PCR screen for positive mCherry cassette integrants. An amplicon of 1400 bp is consistent with correct chromosomal integration. Lane 1: 1 kb Plus Opti-DNA Marker. Lane 2-6: Colonies #1-5.
B) Confirmation of mCherry Integration
- Positive screened colonies were grown in liquid media and pelleted to reveal mCherry expression (Figure 4).
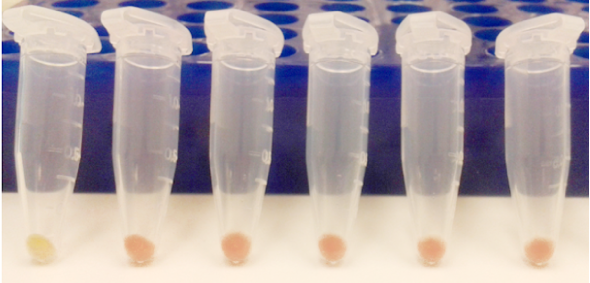
Figure 4. mCherry positive clones express red fluorescent protein thus producing a pink-red phenotype. The wildtype E. coli strain is depicted in the first tube on the left.
C) Sequencing of mCherry Positive Colonies
- PCR products were subjected to Sanger sequencing to confirm correct insertion and knock-in of the mCherry cassette (Figure 5).
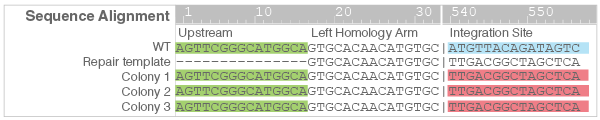
Figure 5. Sequence alignment of mCherry positive colonies compared to wildtype and repair template sequences. The upstream sequence (green) confirms correct integration and a portion of the mCherry cassette sequence (red) is shown to differ from the wildtype sequence (blue).
Related Products

FAQs
How should I submit E. coli cells for this service?
Using sterile technique, grow the specific strain overnight without selection, ensuring that it is turbid and dense the next day. In an appropriate cryotube vial, mix the overnight dense culture with glycerol to a final concentration of 15% (for a total of volume of at least 1ml). Freeze at -80°C, until ready to ship on dry ice to abm.
Citations

